
| 产品编号 | bsm-61024R |
| 英文名称 | Dystrophin Recombinant Rabbit mAb |
| 中文名称 | 肌营养不良蛋白重组兔单抗 |
| 别 名 | BMD; CMD3B; DXS142; DXS164; DXS206; DXS230; DXS239; DXS268; DXS269; DXS270; DXS272; MRX85; DXSmh7; DXSmh9; Dp427; Dp71; dys; mdx; pke; DNADMD1; DMD_HUMAN; DMD; DMD_MOUSE; DMD_RAT; |
| 抗体来源 | Rabbit |
| 克隆类型 | Recombinant |
| 克 隆 号 | 8C6 |
| 交叉反应 | Human,Mouse,Rat |
| 产品应用 | IHC-P=1:50-200,IHC-F=1:50-200,IF=1:50-200
not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 理论分子量 | 405 kDa |
| 细胞定位 | 细胞浆 细胞膜 |
| 性 状 | Liquid |
| 免 疫 原 | A synthesized peptide derived from human Dystrophin: 3611-3685 |
| 亚 型 | IgG |
| 纯化方法 | affinity purified by Protein A |
| 缓 冲 液 | 0.01M TBS(pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol. |
| 保存条件 | Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. |
| 注意事项 | This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| PubMed | PubMed |
| 产品介绍 |
This gene spans a genomic range of greater than 2 Mb and encodes a large protein containing an N-terminal actin-binding domain and multiple spectrin repeats. The encoded protein forms a component of the dystrophin-glycoprotein complex (DGC), which bridges the inner cytoskeleton and the extracellular matrix. Deletions, duplications, and point mutations at this gene locus may cause Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), or cardiomyopathy. Alternative promoter usage and alternative splicing result in numerous distinct transcript variants and protein isoforms for this gene. [provided by RefSeq, Dec 2016]
Function: Anchors the extracellular matrix to the cytoskeleton via F-actin. Ligand for dystroglycan. Component of the dystrophin-associated glycoprotein complex which accumulates at the neuromuscular junction (NMJ) and at a variety of synapses in the peripheral and central nervous systems and has a structural function in stabilizing the sarcolemma. Also implicated in signaling events and synaptic transmission. Subunit: Interacts with SYNM (By similarity). Interacts with the syntrophins SNTA1, SNTB1, SNTB2, SNTG1 and SNTG2. Interacts with KRT19. Component of the dystrophin-associated glycoprotein complex which is composed of three subcomplexes: a cytoplasmic complex comprised of DMD (or UTRN), DTNA and a number of syntrophins, such as SNTB1, SNTB2, SNTG1 and SNTG2, the transmembrane dystroglycan complex, and the sarcoglycan-sarcospan complex. Interacts with DAG1 (betaDAG1) with DMD; the interaction is inhibited by phosphorylation on the PPXY motif of DAG1. Subcellular Location: Cell membrane; sarcolemma. Cytoplasm; cytoskeleton. Tissue Specificity: Expressed in muscle fibers accumulating in the costameres of myoplasm at the sarcolemma. Expressed in brain, muscle, kidney, lung and testis. Isoform 5 is expressed in heart, brain, liver, testis and hepatoma cells. Most tissues contain transcripts of multiple isoforms, however only isoform 5 is detected in heart and liver. DISEASE: Defects in DMD are the cause of Duchenne muscular dystrophy (DMD) [MIM:310200]. DMD is the most common form of muscular dystrophy; a sex-linked recessive disorder. It typically presents in boys aged 3 to 7 year as proximal muscle weakness causing waddling gait, toe-walking, lordosis, frequent falls, and difficulty in standing up and climbing up stairs. The pelvic girdle is affected first, then the shoulder girdle. Progression is steady and most patients are confined to a wheelchair by age of 10 or 12. Flexion contractures and scoliosis ultimately occur. About 50% of patients have a lower IQ than their genetic expectations would suggest. There is no treatment. Defects in DMD are the cause of Becker muscular dystrophy (BMD) [MIM:300376]. BMD resembles DMD in hereditary and clinical features but is later in onset and more benign. Defects in DMD are a cause of cardiomyopathy dilated X-linked type 3B (CMD3B) [MIM:302045]; also known as X-linked dilated cardiomyopathy (XLCM). Dilated cardiomyopathy is a disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. Similarity: Contains 2 CH (calponin-homology) domains. Contains 22 spectrin repeats. Contains 1 WW domain. Contains 1 ZZ-type zinc finger. SWISS: P11532 Gene ID: 1756 Database links: Entrez Gene: 1756 Human Entrez Gene: 13405 Mouse Omim: 300377 Human SwissProt: P11532 Human SwissProt: P11531 Mouse Unigene: 495912 Human Unigene: 275608 Mouse Unigene: 416750 Mouse Unigene: 10307 Rat |
| 产品图片 |
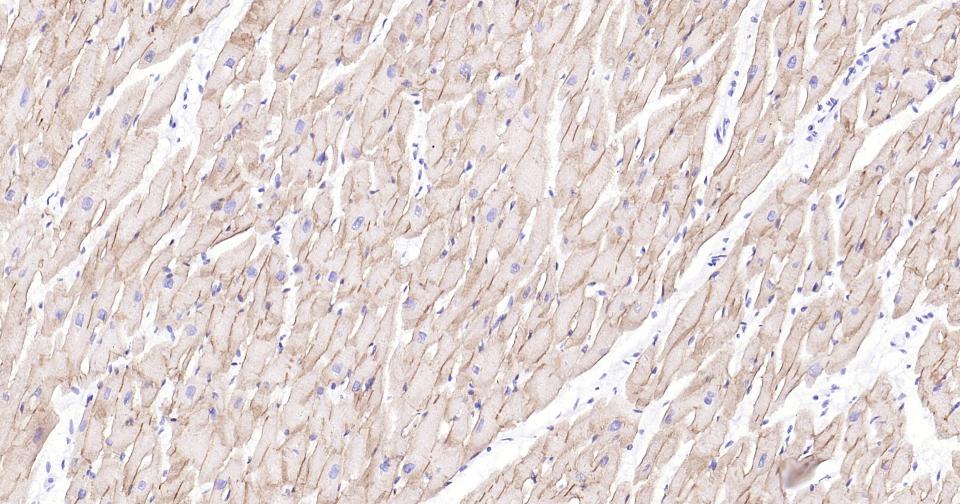
Paraformaldehyde-fixed, paraffin embedded Human Heart; Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15 min; Antibody incubation with Dystrophin Monoclonal Antibody, Unconjugated(bsm-61024R) at 1:100 overnight at 4°C, followed by conjugation to the SP Kit (Rabbit, SP-0023) and DAB (C-0010) staining.
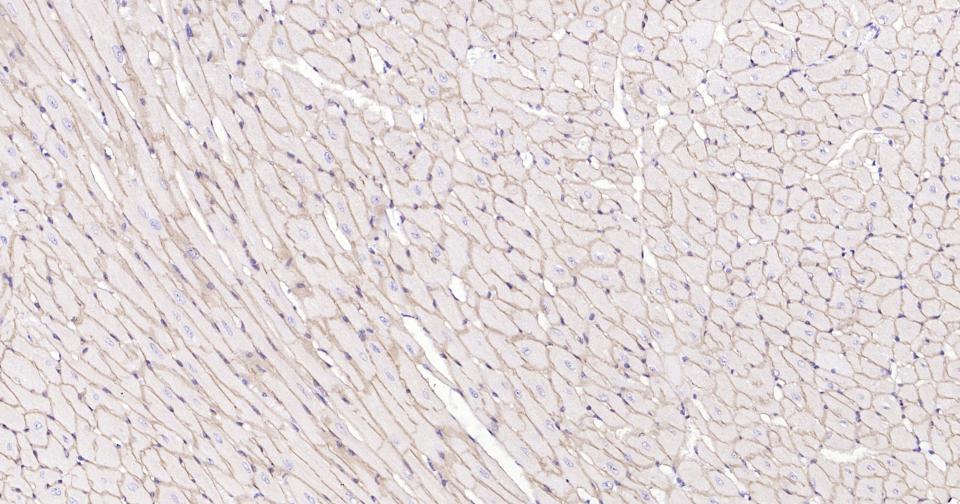
Paraformaldehyde-fixed, paraffin embedded Mouse Heart; Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15 min; Antibody incubation with Dystrophin Monoclonal Antibody, Unconjugated(bsm-61024R) at 1:100 overnight at 4°C, followed by conjugation to the SP Kit (Rabbit, SP-0023) and DAB (C-0010) staining.
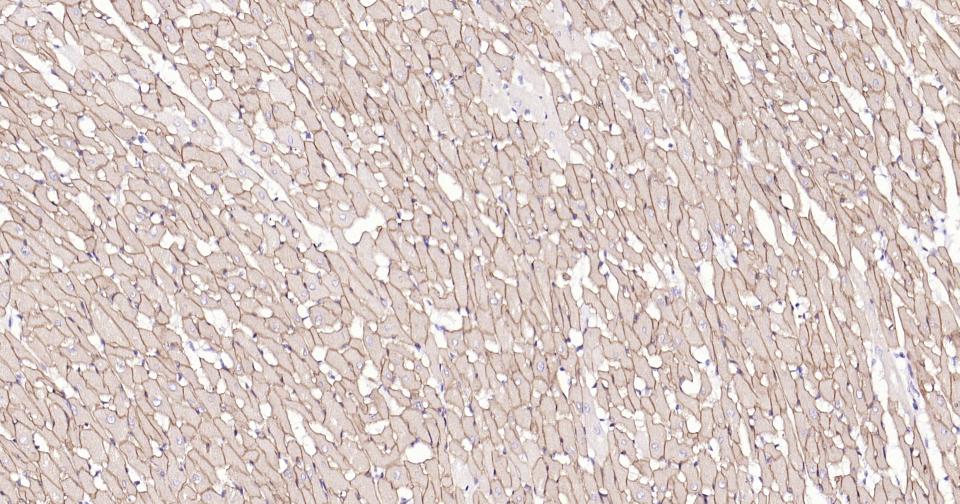
Paraformaldehyde-fixed, paraffin embedded Rat Heart; Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15 min; Antibody incubation with Dystrophin Monoclonal Antibody, Unconjugated(bsm-61024R) at 1:100 overnight at 4°C, followed by conjugation to the SP Kit (Rabbit, SP-0023) and DAB (C-0010) staining.

Paraformaldehyde-fixed, paraffin embedded Human Skeletal muscle; Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15 min; Antibody incubation with Dystrophin Monoclonal Antibody, Unconjugated(bsm-61024R) at 1:100 overnight at 4°C, followed by conjugation to the SP Kit (Rabbit, SP-0023) and DAB (C-0010) staining.
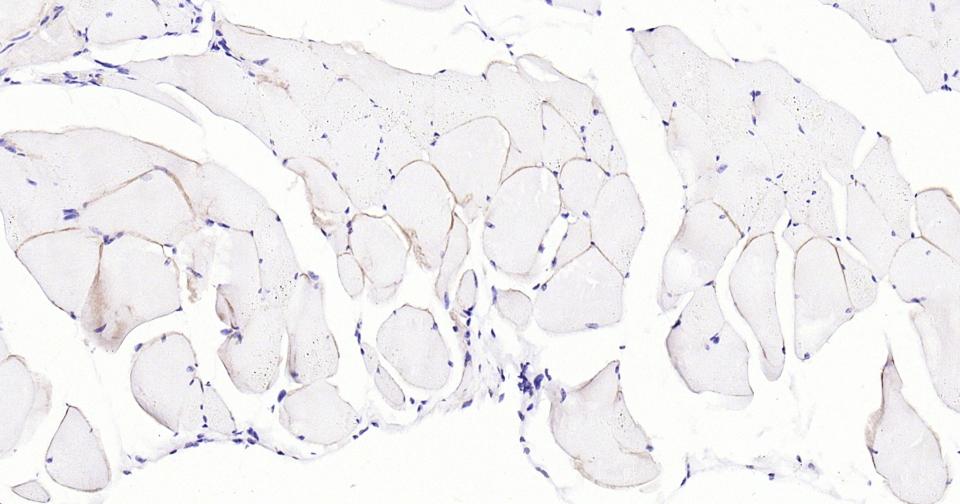
Paraformaldehyde-fixed, paraffin embedded Mouse Skeletal muscle; Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15 min; Antibody incubation with Dystrophin Monoclonal Antibody, Unconjugated(bsm-61024R) at 1:100 overnight at 4°C, followed by conjugation to the SP Kit (Rabbit, SP-0023) and DAB (C-0010) staining.
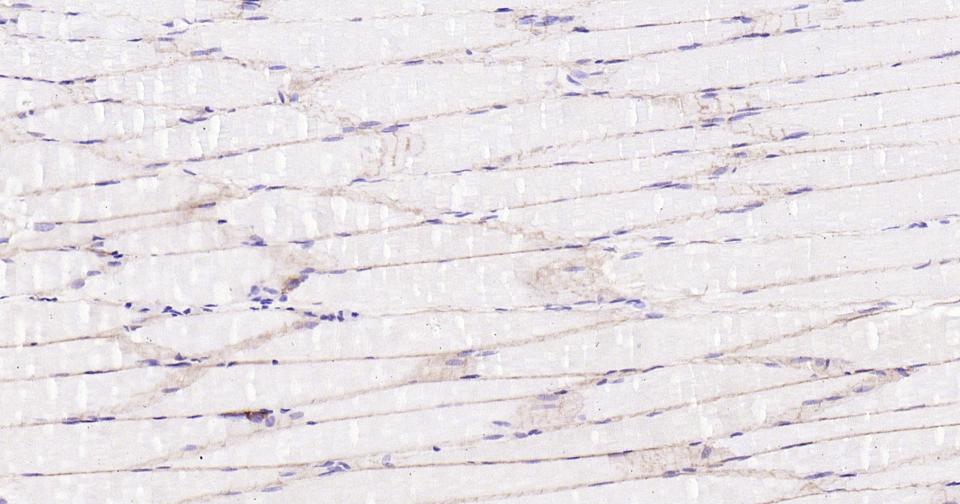
Paraformaldehyde-fixed, paraffin embedded Rat Skeletal muscle; Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15 min; Antibody incubation with Dystrophin Monoclonal Antibody, Unconjugated(bsm-61024R) at 1:100 overnight at 4°C, followed by conjugation to the SP Kit (Rabbit, SP-0023) and DAB (C-0010) staining.
|
| 1、抗体溶解方法 | |
| 2、抗体修复方式 | |
| 3、常用试剂的配制 | |
| 4、免疫组化操作步骤 | |
| 5、免疫组化问题解答 | |
| 6、Western Blotting 操作步骤 | |
| 7、Western Blotting 问题解答 | |
| 8、关于肽链的设计 | |
| 9、多肽的溶解与保存 | |
| 10、酶标抗体效价测定程序 | |




