

| 产品编号 | bs-13858R |
| 英文名称 | CEP290 Rabbit pAb |
| 中文名称 | 中心体蛋白290抗体 |
| 别 名 | 3H11Ag; BBS14; CT87; JBTS5; LCA10; MKS4; NPHP6; POC3; SLSN6; rd16; CE290_HUMAN; CEP290; Bardet-Biedl syndrome 14 protein; Cancer/testis antigen 87 (CT87); Nephrocystin-6; Tumor antigen se2-2; KIAA0373; |
| 研究领域 | 细胞生物 神经生物学 信号转导 表观遗传学 |
| 抗体来源 | Rabbit |
| 克隆类型 | Polyclonal |
| 克 隆 号 | |
| 交叉反应 | Mouse (predicted: Human,Rat,Sheep,Cow,Dog,Horse) |
| 产品应用 | IHC-P=1:100-500,IHC-F=1:100-500,IF=1:100-500
not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 理论分子量 | 291 kDa |
| 细胞定位 | 细胞浆 |
| 性 状 | Liquid |
| 浓 度 | 1mg/ml |
| 免 疫 原 | KLH conjugated synthetic peptide derived from human CEP290: 131-230/2479 |
| 亚 型 | IgG |
| 纯化方法 | affinity purified by Protein A |
| 缓 冲 液 | 0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol. |
| 保存条件 | Shipped at 4℃. Store at -20℃ for one year. Avoid repeated freeze/thaw cycles. |
| 注意事项 | This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| PubMed | PubMed |
| 产品介绍 |
This gene encodes a protein with 13 putative coiled-coil domains, a region with homology to SMC chromosome segregation ATPases, six KID motifs, three tropomyosin homology domains and an ATP/GTP binding site motif A. The protein is localized to the centrosome and cilia and has sites for N-glycosylation, tyrosine sulfation, phosphorylation, N-myristoylation, and amidation. Mutations in this gene have been associated with Joubert syndrome and nephronophthisis and the presence of antibodies against this protein is associated with several forms of cancer. [provided by RefSeq, Jul 2008 Function: Activates ATF4-mediated transcription. Required for the correct localization of ciliary and phototransduction proteins in retinal photoreceptor cells; may play a role in ciliary transport processes. Subcellular Location: Cytoplasm > cytoskeleton > centrosome. Nucleus. Cell projection > cilium. Connecting cilium of photoreceptor cells, base of cilium in kidney intramedullary collecting duct cells. Tissue Specificity: Ubiquitous. Expressed strongly in placenta and weakly in brain. DISEASE: Defects in CEP290 are a cause of Joubert syndrome type 5 (JBTS5) [MIM:610188]. Joubert syndrome is an autosomal recessive disease characterized by cerebellar vermis hypoplasia with prominent superior cerebellar peduncles (the 'molar tooth sign' on axial magnetic resonance imaging), psychomotor delay, hypotonia, ataxia, oculomotor apraxia and neonatal breathing abnormalities. JBTS5 shares the neurologic and neuroradiologic features of Joubert syndrome together with severe retinal dystrophy and/or progressive renal failure characterized by nephronophthisis. Defects in CEP290 are a cause of Senior-Loken syndrome type 6 (SLSN6) [MIM:610189]. Senior-Loken syndrome is also known as juvenile nephronophthisis with Leber amaurosis. It is an autosomal recessive renal-retinal disorder, characterized by progressive wasting of the filtering unit of the kidney, with or without medullary cystic renal disease, and progressive eye disease. Defects in CEP290 are the cause of Leber congenital amaurosis type 10 (LCA10) [MIM:611755]. LCA designates a clinically and genetically heterogeneous group of childhood retinal degenerations, generally inherited in an autosomal recessive manner. Affected infants have little or no retinal photoreceptor function as tested by electroretinography. LCA represents the most common genetic cause of congenital visual impairment in infants and children. Defects in CEP290 are the cause of Meckel syndrome type 4 (MKS4) [MIM:611134]. MKS4 is an autosomal recessive disorder characterized by a combination of renal cysts and variably associated features including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia and cysts, and polydactyly. Note=Antibodies against CEP290 are present in sera from patients with cutaneous T-cell lymphomas, but not in the healthy control population. Defects in CEP290 are the cause of Bardet-Biedl syndrome type 14 (BBS14) [MIM:209900]. A syndrome characterized by usually severe pigmentary retinopathy, early-onset obesity, polydactyly, hypogenitalism, renal malformation and mental retardation. Secondary features include diabetes mellitus, hypertension and congenital heart disease. Inheritance is autosomal recessive, but three mutated alleles (two at one locus, and a third at a second locus) may be required for disease manifestation in some cases (triallelic inheritance). SWISS: O15078 Gene ID: 80184 Database links: Entrez Gene: 80184 Human Omim: 610142 Human wissProt: O15078 Human Unigene: 150444 Human |
| 产品图片 |
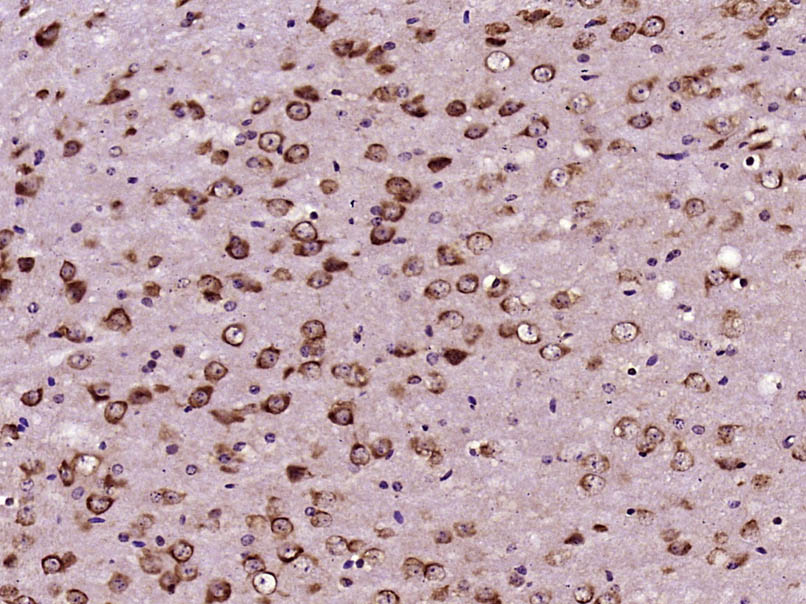
Paraformaldehyde-fixed, paraffin embedded (Mouse brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (CEP290) Polyclonal Antibody, Unconjugated (bs-13858R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
|
| 1、抗体溶解方法 | |
| 2、抗体修复方式 | |
| 3、常用试剂的配制 | |
| 4、免疫组化操作步骤 | |
| 5、免疫组化问题解答 | |
| 6、Western Blotting 操作步骤 | |
| 7、Western Blotting 问题解答 | |
| 8、关于肽链的设计 | |
| 9、多肽的溶解与保存 | |
| 10、酶标抗体效价测定程序 | |




