

| 产品编号 | bs-3955R |
| 英文名称 | MT-ND6 Rabbit pAb |
| 中文名称 | NADH复合体6抗体 |
| 别 名 | MTND6; NU6M_HUMAN; MT-ND6; NADH dehydrogenase subunit 6; 7.1.1.2; NADH6; ND6; NU6M_PIG; mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 6; NADH dehydrogenase 6; mitochondrially encoded NADH dehydrogenase 6; complex I ND6 subunit; NADH-ubiquinone oxidoreductase chain 6 |
 |
Specific References (3) | bs-3955R has been referenced in 3 publications.
[IF=15.84] Ke Cao. et al. Hypermethylation of Hepatic Mitochondrial ND6 Provokes Systemic Insulin Resistance. 2021 May 02 WB ; Mouse.
[IF=5.2] Wu, Ji-hong, et al. "Cumulative mtDNA damage and mutations contribute to the progressive loss of RGCs in a rat model of glaucoma." Neurobiology of Disease (2014). WB ; Rat.
[IF=0] Hoque SAM et al. Mitochondrial protein turnover is critical for granulosa cell proliferation and differentiation in antral follicles. J Endocr Soc. 2018 Dec 10;3(2):324-339. WB ; Mouse.
|
| 研究领域 | 肿瘤 细胞生物 免疫学 神经生物学 转录调节因子 |
| 抗体来源 | Rabbit |
| 克隆类型 | Polyclonal |
| 交叉反应 | Human,Rat (predicted: Mouse,Sheep,Cow,Dog,Horse) |
| 产品应用 | IHC-P=1:100-500,IHC-F=1:100-500,IF=1:100-500,Flow-Cyt=1ug/Test
not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 理论分子量 | 19kDa |
| 细胞定位 | 细胞浆 |
| 性 状 | Liquid |
| 浓 度 | 1mg/ml |
| 免 疫 原 | KLH conjugated synthetic peptide derived from human MTND6: 51-150/174 |
| 亚 型 | IgG |
| 纯化方法 | affinity purified by Protein A |
| 缓 冲 液 | 0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol. |
| 保存条件 | Shipped at 4℃. Store at -20℃ for one year. Avoid repeated freeze/thaw cycles. |
| 注意事项 | This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| PubMed | PubMed |
| 产品介绍 |
NADH Dehydrogenase subunit 6 (MTND6) is 1 of the 7 mitochondrial DNA (mtDNA) encoded subunits (MTND1, MTND2, MTND3, MTND4L, MTND4, MTND5, MTND6) included among the approximately 41 polypeptides of respiratory Complex I. Complex I accepts electrons from NADH, transfers them to ubiquinone (Coenzyme Q10), and uses the energy released to pump protons across the mitochondria inner membrane. MTND6 has been proposed to be a component of the iron-protein fragment. Function: Core subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase (Complex I) that is believed to belong to the minimal assembly required for catalysis. Complex I functions in the transfer of electrons from NADH to the respiratory chain. The immediate electron acceptor for the enzyme is believed to be ubiquinone (By similarity). Subcellular Location: Mitochondrion membrane; Multi-pass membrane protein (Potential). DISEASE: Defects in MT-ND6 are a cause of Leber hereditary optic neuropathy (LHON) [MIM:535000]. LHON is a maternally inherited disease resulting in acute or subacute loss of central vision, due to optic nerve dysfunction. Cardiac conduction defects and neurological defects have also been described in some patients. LHON results from primary mitochondrial DNA mutations affecting the respiratory chain complexes. Defects in MT-ND6 are a cause of Leber hereditary optic neuropathy with dystonia (LDYT) [MIM:500001]; also called familial dystonia with visual failure and striatal lucencies. LDYT is part of a spectrum of Leber hereditary optic neuropathy. It is characterized by the association of optic atrophy and central vision loss with dystonia. Defects in MT-ND6 are a cause of mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes syndrome (MELAS) [MIM:540000]. MELAS is a genetically heterogenious disorder, characterized by episodic vomiting, seizures, and recurrent cerebral insults resembling strokes and causing hemiparesis, hemianopsia, or cortical blindness. Defects in MT-ND6 are a cause of mitochondrial complex I deficiency (MT-C1D) [MIM:252010]. A disorder of the mitochondrial respiratory chain that causes a wide range of clinical manifestations from lethal neonatal disease to adult-onset neurodegenerative disorders. Phenotypes include macrocephaly with progressive leukodystrophy, non-specific encephalopathy, cardiomyopathy, myopathy, liver disease, Leigh syndrome, Leber hereditary optic neuropathy, and some forms of Parkinson disease. Similarity: Belongs to the complex I subunit 6 family. SWISS: P03923 Gene ID: 4541 Database links: Entrez Gene: 4541 Human Omim: 516006 Human SwissProt: P03923 Human |
| 产品图片 |
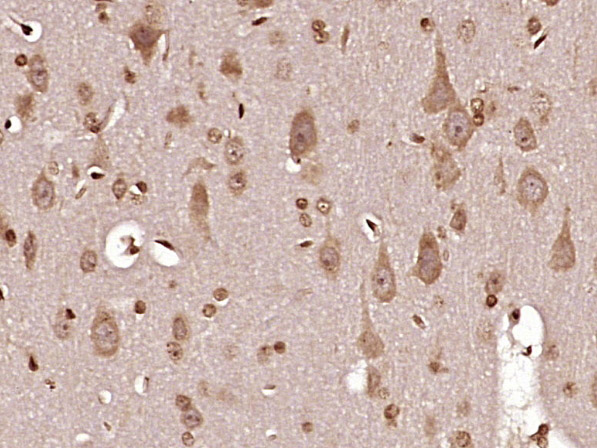
Paraformaldehyde-fixed, paraffin embedded (rat brain tissue); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (MT-ND6) Polyclonal Antibody, Unconjugated (bs-3955R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
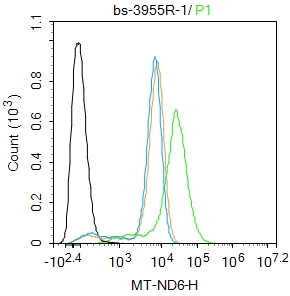
Blank control: THP-1.
Primary Antibody (green line): Rabbit Anti-MT-ND6 antibody (bs-3955R)
Dilution: 1μg /10^6 cells;
Isotype Control Antibody (orange line): Rabbit IgG .
Secondary Antibody : Goat anti-rabbit IgG-FITC
Dilution: 0.5μg /test.
Protocol
The cells were fixed with 4% PFA (10min at room temperature)and then permeabilized with 0.1% PBST for 20 min at room temperature. The cells were then incubated in 5%BSA to block non-specific protein-protein interactions for 30 min at room temperature .Cells stained with Primary Antibody for 30 min at room temperature. The secondary antibody used for 40 min at room temperature. Acquisition of 20,000 events was performed.
|
| 1、抗体溶解方法 | |
| 2、抗体修复方式 | |
| 3、常用试剂的配制 | |
| 4、免疫组化操作步骤 | |
| 5、免疫组化问题解答 | |
| 6、Western Blotting 操作步骤 | |
| 7、Western Blotting 问题解答 | |
| 8、关于肽链的设计 | |
| 9、多肽的溶解与保存 | |
| 10、酶标抗体效价测定程序 | |





