

| 产品编号 | bs-0235R |
| 英文名称 | PMP22 Rabbit pAb |
| 中文名称 | 外周髓鞘蛋白-22抗体 |
| 别 名 | CIDP; CMT1A; CMT1E; DSS; GAS-3; GAS3; HMSNIA; HNPP; Sp110; PMP22_HUMAN; PMP22; PMP-22; Growth arrest-specific protein 3 (GAS-3); PMP22_RAT; Protein CD25; SR13 myelin protein; Schwann cell membrane glycoprotein (SAG); Cd25; PXMP2_RAT; Pxmp2; 22 kDa peroxis |
 |
Specific References (1) | bs-0235R has been referenced in 1 publications.
[IF=0] H Xuan et al. Freestanding Hyaluronic Acid/Silk-based Self-healing Coating towards Tissue Repair with Antibacterial Surface. ACS Appl. Bio Mater., (Web): 28 Jan 2020. WB ; Rat.
|
| 研究领域 | 免疫学 神经生物学 糖蛋白 |
| 抗体来源 | Rabbit |
| 克隆类型 | Polyclonal |
| 克 隆 号 | |
| 交叉反应 | Mouse,Rat (predicted: Human) |
| 产品应用 | WB=1:500-2000
not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 理论分子量 | 22 kDa |
| 检测分子量 | 27 |
| 细胞定位 | 细胞膜 |
| 性 状 | Liquid |
| 浓 度 | 1mg/ml |
| 免 疫 原 | KLH conjugated synthetic peptide derived from human PMP22: 101-160/160 <Extracellular> |
| 亚 型 | IgG |
| 纯化方法 | affinity purified by Protein A |
| 缓 冲 液 | 0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol. |
| 保存条件 | Shipped at 4℃. Store at -20℃ for one year. Avoid repeated freeze/thaw cycles. |
| 注意事项 | This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| PubMed | PubMed |
| 产品介绍 |
PMP22 is a 22 kDa glycoprotein expressed in the compact myelin of the peripheral nervous system. In the peripheral nervous system, PMP 22 is produced by myelinating Schwann cells and is coexpressed with the genes for myelin basic protein (MBP) during nerve development and regeneration. Alterations in the level of this protein cause several genetic human diseases. If the protein is duplicated, patients develop Charcot Marie Tooth disease. If one copy of the gene is deleted, they suffer from the inherited tendency to pressure palsies. Function: Might be involved in growth regulation, and in myelinization in the peripheral nervous system. Subcellular Location: Cell membrane; Multi-pass membrane protein. DISEASE: Defects in PMP22 are the cause of Charcot-Marie-Tooth disease type 1A (CMT1A) [MIM:118220]; also known as hereditary motor and sensory neuropathy IA. CMT1A is a form of Charcot-Marie-Tooth disease, the most common inherited disorder of the peripheral nervous system. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathy or CMT1, and primary peripheral axonal neuropathy or CMT2. Neuropathies of the CMT1 group are characterized by severely reduced nerve conduction velocities (less than 38 m/sec), segmental demyelination and remyelination with onion bulb formations on nerve biopsy, slowly progressive distal muscle atrophy and weakness, absent deep tendon reflexes, and hollow feet. CMT1A inheritance is autosomal dominant. Defects in PMP22 are a cause of Dejerine-Sottas syndrome (DSS) [MIM:145900]; also known as Dejerine-Sottas neuropathy (DSN) or hereditary motor and sensory neuropathy III (HMSN3). DSS is a severe degenerating neuropathy of the demyelinating Charcot-Marie-Tooth disease category, with onset by age 2 years. DSS is characterized by motor and sensory neuropathy with very slow nerve conduction velocities, increased cerebrospinal fluid protein concentrations, hypertrophic nerve changes, delayed age of walking as well as areflexia. There are both autosomal dominant and autosomal recessive forms of Dejerine-Sottas syndrome. Defects in PMP22 are a cause of hereditary neuropathy with liability to pressure palsies (HNPP) [MIM:162500]; an autosomal dominant disorder characterized by transient episodes of decreased perception or peripheral nerve palsies after slight traction, compression or minor traumas. Defects in PMP22 are the cause of Charcot-Marie-Tooth disease type 1E (CMT1E) [MIM:118300]; also known as Charcot-Marie-Tooth disease and deafness autosomal dominant. CMT1E is an autosomal dominant form of Charcot-Marie-Tooth disease characterized by the association of sensorineural hearing loss with peripheral demyelinating neuropathy. Defects in PMP22 may be a cause of inflammatory demyelinating polyneuropathy (IDP) [MIM:139393]. IDP is a putative autoimmune disorder presenting in an acute (AIDP) or chronic form (CIDP). The acute form is also known as Guillain-Barre syndrome. Similarity: Belongs to the PMP-22/EMP/MP20 family. SWISS: Q01453 Gene ID: 5376 Database links: Entrez Gene: 5376 Human Omim: 601097 Human SwissProt: Q01453 Human Unigene: 372031 Human Unigene: 1476 Rat 神经生物学相关蛋白(Neurobiology) 外周髓鞘蛋白22(PMP22)是一种糖蛋白,在外周神经系统中的致密肌纤维素中表达。它由髓鞘雪旺氏细胞产生,并在神经发育和再生过程中与MBP和Po蛋白共同表达。该蛋白表达水平变化可引起几种人类遗传性疾病,如果该蛋白增多,则病人会发生Charcot-Marie-Tooth疾病,如果缺失,则易患压力麻痹的遗传倾向。 |
| 产品图片 |
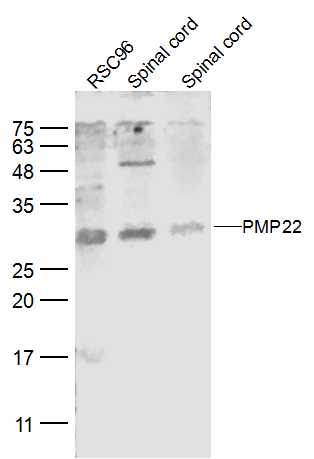
Sample:
RSC96 (Mouse) CellLysate at 30 ug
Spinal cord (Mouse) Lysate at 40 ug
Spinal cord (Rat) Lysate at 40 ug
Primary: Anti-PMP22 (bs-0235R) at 1/500 dilution
Secondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilution
Predicted band size: 22 kD
Observed band size: 27 kD
|
| 1、抗体溶解方法 | |
| 2、抗体修复方式 | |
| 3、常用试剂的配制 | |
| 4、免疫组化操作步骤 | |
| 5、免疫组化问题解答 | |
| 6、Western Blotting 操作步骤 | |
| 7、Western Blotting 问题解答 | |
| 8、关于肽链的设计 | |
| 9、多肽的溶解与保存 | |
| 10、酶标抗体效价测定程序 | |





